Es werden nicht nur Graphen-Kohlenstoff-Nanoröhren eingeführt, sondern auch neue Kohlenstoff-Nanomaterialien und ihre Hilfsmechanismen.
Fulleren, Kohlenstoffnanoröhren (CNTs, Carbon Nanotubes) und Graphene (Graphene) sind in den letzten Jahren beliebte Kohlenstoffnanomaterialien. Derzeit haben fünf Wissenschaftler auf diesem Gebiet den Nobelpreis erhalten. Warum sind Kohlenstoffnanomaterialien sehr gefragt? Beispielsweise machen Fahrräder aus Stahl mit Kohlenstofffaserzusatz nur einen Bruchteil des Gewichts von gewöhnlichen Fahrrädern aus, da die Masse der Kohlenstoffatome sehr gering ist und die chemischen Bindungen zwischen Kohlenstoffatomen oder zwischen Kohlenstoffatomen und anderen Atomen bestehen. Sehr stark. Daher weisen mit Kohlenstoffnanometern gemischte Materialien normalerweise bessere mechanische Eigenschaften und ein geringeres Gesamtgewicht auf.
Erste Prinzipien sind in der Physik, Chemie und Materialwissenschaft weit verbreitet. Materialdesign, Materialvorhersage, Interpretationsexperimente usw. sind untrennbar mit der Berechnung der ersten Prinzipien verbunden, da das erste Prinzip von der Schrödinger-Gleichung ausgeht und nur sehr wenige Parameter benötigt, um die meisten Materialeigenschaften des Materials sehr genau zu berechnen. In Kombination mit der adiabatischen Annahme kann damit auch die Molekulardynamik simuliert werden. Auf dem Gebiet der Kohlenstoffnanomaterialien werden häufig First-Principles-Berechnungen verwendet, da die elektronische Korrelation von Kohlenstoffatomen sehr schwach ist und die First-Principles-Berechnungen häufig sehr genaue Vorhersagen treffen können.
In diesem Artikel werden einige neue Arten von Kohlenstoff-Nanomaterialien vorgestellt, die sich in der Art und Weise, wie Kohlenstoffatome kombiniert und in bekannten Fullerenen, Kohlenstoff-Nanoröhren und Graphen angeordnet werden, geringfügig unterscheiden. Diese subtilen Unterschiede können sich in den endgültigen Materialeigenschaften widerspiegeln, jedoch stark variieren. Ein kleiner Unterschied in der Anordnung der Kohlenstoffatome kann zu großen Unterschieden in den Materialeigenschaften führen. Hier ziehen Kohlenstoffnanomaterialien viele Materialwissenschaftler, Physiker und Chemiker an.
1. Hybridisierung und Dimension
Es gibt zwei Hauptmethoden, um Kohlenstoffatome an Kohlenstoffnanomaterialien zu hybridisieren: sp2 oder sp3. Im sp2-Hybridmodus bildet jedes Kohlenstoffatom drei Molekülorbitale, die gleichmäßig in einer Ebene in einem Winkel von 120 Grad verteilt sind, und einen außerhalb der Ebene liegenden p-Orbit, der gemeinhin als pz-Orbital bezeichnet wird. Die typischsten Kohlenstoffnanomaterialien Es ist ein berühmtes Graphen. Im sp3-Hybridmodus bildet jedes Kohlenstoffatom vier Molekülorbitale, die gleichmäßig im Raum verteilt sind und ungefähr die Form eines regelmäßigen Tetraeders vom Körper zu den vier Eckpunkten bilden. Ein typischer Feststoff repräsentiert einen Diamanten, ein typischer Vertreter der Welt der Nanomaterialien ist Adamantan. Adamantan ist ein Vertreter einer ganzen Familie von Materialien, und ein Molekül enthält einen Kern der Diamantstruktur. Wenn es mehrere Kerne mit Diamantstruktur enthält, wird diese Materialfamilie zu Diamantoid. Abbildung 1: Typische Kohlenstoffnanomaterialien, klassifiziert nach Hybridisierung (sp2, erste Reihe oder sp3, zweite Reihe) und Materialdimensionen.
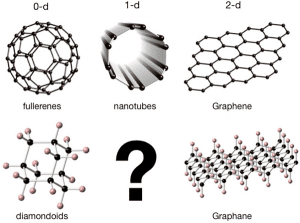
Das Obige ist nur eine Hybridisierung oder vielmehr eine Hauptentscheidung, die ein einzelnes Kohlenstoffatom bei der Bildung eines Nanomaterials treffen kann. Wenn zusätzlich zur Hybridisierung viele Kohlenstoffatome kombiniert werden, können sie sich beliebig ausdehnen. Ist es ein nulldimensionales Material oder ein Material mit hohem Breitengrad? Die obige Tabelle 1 listet verschiedene repräsentative Materialien nach Hybridisierung und Dimension auf.
Eindimensionale Materialien im sp3-Hybridmodus weisen kein typisches Merkmal auf. Leser, die mit einschlägigen Forschungen vertraut sind, mögen an Polyethylen denken, aber in Bezug auf einzelne Moleküle fehlt es Polyethylenmolekülen an einigen Regeln für die Konfiguration über große Entfernungen oder an der Reihenfolge über große Entfernungen, und es fehlt ihnen das Verlangen nach Kohlenstoffnanomaterialien. Mechanische Festigkeit.
2. Kohlenstoffnanodrähte
Ist das Material unten ein bisschen interessant? Ist es fest oder Makromolekül?
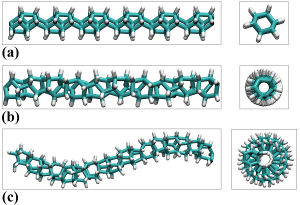
Diese neue Art von Kohlenstoffnanomaterial ist sowohl ein sp3-Hybrid aus Kohlenstoffatomen als auch eine eindimensionale Zusammensetzung von Kohlenstoffatomen. Gleichzeitig sind ihre Querschnitte nicht wie bei einem herkömmlichen linearen organischen Molekül, sondern weisen mehrere chemische Bindungen auf. Durchqueren Sie den Querschnitt. Dies bedeutet, dass diese Materialien hinsichtlich der elektronischen Eigenschaften in der Nähe von Diamantisolatoren liegen. Sie sind herkömmlichen linearen organischen Molekülen in ihren mechanischen Eigenschaften weit überlegen, und ihre mechanische Festigkeit entspricht in etwa der von Kohlenstoffnanoröhren oder Graphen. Theoretische Berechnungen bestätigen dies [1], sie werden Kohlenstoffnanodrähte oder Diamantnanofäden genannt.
Ist dieses neue Material mit einer seltsamen Form nur eine theoretische Erwartung oder kann es tatsächlich hergestellt werden? Es scheint, dass solche Materialien von der Synthese kleiner organischer Moleküle nach einem kleinen bis großen Prozess ausgehen müssen, experimentell [2] jedoch durch einen Prozess von groß nach klein, ausgehend vom festen Zustand von Benzol, nach 25GPa Hochdruck Die Rolle der ursprünglichen sp2-Hybridbindung wird unter hohem Druck zu einer sp3-Hybridbindung, wodurch der dreidimensionale Molekülkristall in ein eindimensionales Kohlenstoffnanomaterial umgewandelt wird.
Im Beispiel von Abbildung 2 sind eindimensionale Nanodrähte mit großer Reichweite dargestellt. In tatsächlichen Experimenten können oft ungeordnete Strukturen erhalten werden. Diese Abbildung zeigt eine ungeordnete Struktur und die Ergebnisse der Rastertunnelmikroskopie von Kohlenstoffnanodrahtkristallen, die in Experimenten erhalten wurden.
3.Anwenden von First-Principles-Berechnungen
First-Principles-Berechnungen liefern gute Ergebnisse bei der Vorhersage der Materialeigenschaften. Die Kombination experimenteller Ergebnisse führt häufig zu tiefergehenden Perspektiven bei der Interpretation experimenteller Ergebnisse. Bei der Synthese von Diamant-Kohlenstoff-Nanodrähten muss aufgrund der harten experimentellen Bedingungen der hohe Druck von 25 GPa in einer sehr kleinen Diamant-Amboss-Zelle (DAC) realisiert werden, sodass der experimentellen Synthese von Materialien experimentelle Ergebnisse mit langer Ordnung fehlen Auf den ersten Blick gibt es eine Menge Störungen. Die theoretischen Berechnungen können uns helfen, zu unterscheiden, ob die Zusammensetzung die neuen Materialien enthält, die wir erwarten.
Theoretisch sind wir zu einer Kohlenstoffnanodrahtstruktur geworden. Nach dem Hinzufügen einer bestimmten Störung durch Einführung der chemischen Rotation der Stein-Wales-Bindung können wir die theoretische Berechnung verwenden, um die atomare Positionsrelaxation durchzuführen und dann die optimale Struktur mit der niedrigsten Energie zu erhalten. Genaue theoretische Berechnungen können den Abstand zwischen Atomen in einem Material angeben oder die radiale Verteilungsfunktion in einem Material berechnen. Der Vergleich der theoretischen Ergebnisse mit den experimentellen Ergebnissen in 4 bestätigt nicht nur, dass die experimentelle Zusammensetzung mit der theoretischen Struktur übereinstimmt, sondern auch, welche Atomstrukturen der Peakauflösung der experimentellen Ergebnisse entsprechen.
Figure 4. Vergleich der radialen Verteilungsfunktion (RDF) experimentell synthetisierter Nanodrähte mit der simulierten radialen Verteilungsfunktion theoretisch erzeugter Kohlenstoffnanodrahtstrukturen.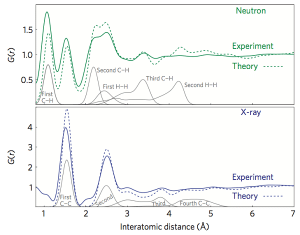
Die erste Prinzipberechnung gibt die optischen Eigenschaften des Materials an. Die Raman-Spektroskopie ist häufig ein zuverlässiges Mittel zur Charakterisierung experimenteller Zusammensetzungen, da sie die experimentelle Zusammensetzung nicht zerstören muss und spektrale Peaks Aufschluss darüber geben können, welche molekularen Schwingungsmoden Raman-Aktivität haben. Eine Methode zur Berechnung des Raman-Spektrums durch die Dichtefunktionaltheorie besteht darin, zuerst die Dielektrizitätskonstante des Moleküls zu berechnen und dann eine kleine Verschiebung der Atomposition entlang des Eigenmodus der Molekülschwingung durchzuführen, um die Änderung der Dielektrizitätskonstante zu berechnen. Mit der fortschrittlichen Rechenleistung moderner Computer können wir nun leicht die Raman-Aktivität eines Moleküls berechnen, um festzustellen, welche Struktureinheiten in der experimentellen Zusammensetzung vorhanden sind. 5 zeigt eine charakteristische Struktureinheit, die in den Syntheseresultaten von Kohlenstoffnanodrähten durch Berechnung und Analyse der Raman-Spektroskopie enthalten ist.
Figure 5. Vergleich experimenteller Raman-Spektren von Kohlenstoffnanodrähten mit der Theorie.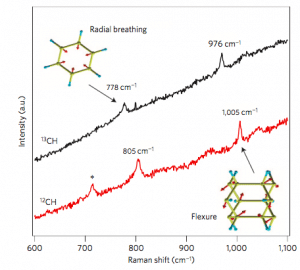
4. Funktionalisierung
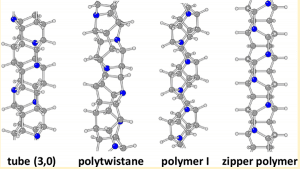
Ein wichtiges Merkmal von Kohlenstoffnanomaterialien ist die Fähigkeit, ihnen verschiedene funktionelle Gruppen hinzuzufügen. Solange einige kleine organische Moleküle in der Herstellungsstufe der synthetischen Zubereitung ersetzt werden. In dem Kohlenstoffnanodrahtmaterial beinhaltet ein einfaches Verfahren das Ersetzen des Wasserstoffatoms (H) in dem Reaktanten durch ein Chloratom (Cl) oder das Ersetzen des Kohlenstoffatoms darin durch ein Stickstoffatom (N) und ein Boratom (B). Es kann funktionalisiert werden, um seine elektronischen Eigenschaften, Phononeigenschaften, thermischen Eigenschaften oder mechanischen Eigenschaften zu ändern. Abbildung 6 zeigt einige typische Nanodrahtstrukturen, die durch Ersetzen von Kohlenwasserstoffgruppen durch Stickstoffatome gebildet werden [4].
Die Untersuchung, Benzol durch einen anfänglichen Reaktanten zu ersetzen, der ein Stickstoffatom enthält, um Nanodrähte zu synthetisieren, ist in dem Artikel [3] veröffentlicht. Dieser Ersatz ist ein vollständiger Ersatz anstelle der Dotierung, wobei Pyridin (Pyridin, C5NH5) anstelle des Benzolrings zur Teilnahme an der Reaktion verwendet wird. Der Reaktionsprozess ähnelt weiterhin der Verwendung von Diamant-Hochdruckballast, in den der sp2-Hybridkohlenstoff umgewandelt wird sp3 hybrid carbon Und vervollständige die Umwandlung kleiner Moleküle in eindimensionale Materialien.
Nach dem Prinzip der ersten Prinzipien können wir zwei Methoden untersuchen, bei denen das Kohlenstoff-Nanodraht-Material dieser Struktur synthetisiert wird. Eine besteht darin, die Charakterisierungseigenschaften aller Kandidatenstrukturen mit Experimenten wie Raman-Spektroskopie, Röntgenbeugung usw. zu vergleichen. Die andere ist natürlich nach ihrer Energie sortiert. Bei der Berechnung der Energie von Kohlenstoffnanodrähten müssen zunächst deren Molekularstruktur und Periodizität optimiert werden. Dieses eindimensionale Material hat jedoch die Eigenschaft, dass es eine helixförmige Struktur aufweist, was zu einigen Schwierigkeiten bei der Berechnung führt.
Wenn Sie die an beiden Enden abgeschnittenen Makromoleküle ersetzen, muss die Energieberechnung ungenau sein. Wenn Sie periodische Randbedingungen verwenden, wie bestimmen Sie den Helixwinkel? Ein möglicher Trick besteht darin, mehrere Spiralwinkel für die Berechnung auszuwählen [2]. Jeder Winkel ist unterschiedlich, was bedeutet, dass die Länge einer strukturellen Wiederholungsperiode entlang der eindimensionalen Struktur unterschiedlich ist. Nach der Berechnung einer Reihe von verschiedenen Spiralwinkeln wird die durchschnittliche Energie pro Struktureinheit (oder der Durchschnitt pro Atom) erhalten und eine einfache quadratische Regressionsanpassung für den Spiralwinkel durchgeführt. Die implizite Annahme einer quadratischen Regressionsanpassung ist, dass der Effekt zwischen zwei benachbarten Strukturelementen ungefähr federartig ist. Obwohl dies keine vollständig zutreffende Hypothese ist, kann sie dennoch die Hauptkraft zwischen benachbarten Einheiten erfassen, da in Kohlenstoffnanomaterialien kovalente Bindungskräfte zwischen benachbarten Atomen und benachbarten Struktureinheiten verwendet werden. Das Hookesche Frühlingsgesetz ist ungefähr.
Figure 6. Vier typische Diamant-Kohlenstoff-Nanodrähte mit Stickstoffatomen aus der Literatur [4]
5. Mechanische festigkeit
Kohlenstoffnanomaterialien haben viele wunderbare elektrische Eigenschaften, aber jetzt sind sie in ihrer mechanischen Leichtigkeit weit verbreitet: leichte Atome, starke Bindung. Kohlenstoffnanodrähte haben die Grundeinheit von Diamanten. Werden sie auch genug Kraft haben? Einfach gesagt, ja. Wie in Abbildung 7 gezeigt, zeigen die Berechnungen, dass die Kohlenstoffnanodrähte einen Elastizitätsmodul zwischen 800 und 930 GPa aufweisen, der mit natürlichen Diamanten (1220 GPa) vergleichbar ist. Natürlich ist die mechanische Festigkeit dieses eindimensionalen Materials gerichtet. Dies ist sowohl ein Nachteil als auch ein Vorteil: Dieses Material bündelt alle mechanischen Festigkeiten in einer Richtung. Einige stellen sich sogar vor, dass mit diesem Kohlenstoffnanodraht ein Kabel für einen Weltraumaufzug hergestellt werden kann.
Figure 7. Young-Modul von drei verschiedenen Arten von Diamant-Kohlenstoff-Nanodrähten aus Lit. [5].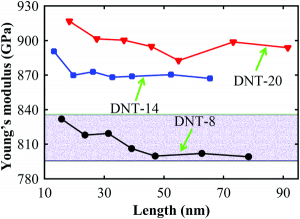
6. Schlussfolgerung
Diamant-Kohlenstoff-Nanodrähte haben sich kürzlich der großen Familie von Kohlenstoff-Nanomaterialien mit einer strengen eindimensionalen Struktur und hoher mechanischer Festigkeit angeschlossen. Im Rahmen des Forschungsprozesses können mithilfe leistungsstarker Rechenleistung mithilfe der First-Principles-Berechnung die mögliche atomare Molekülstruktur von Kohlenstoffnanodrähten untersucht, die Interpretation der experimentellen Ergebnisse unterstützt und die experimentellen Ergebnisse eingehend analysiert werden . Kohlenstoffnanodrähte sowie viele andere interessante neue Merkmale von Kohlenstoffnanostrukturen warten auf weitere theoretische Berechnungen und experimentelle Verifizierungen.
Verweise
1. Fitzgibbons, TC; Guthrie, M .; Xu, E.-s .; Crespi, VH; Davidowski, SK; Cody, GD; Alem, N; Badding, JV Mater. 2014, 14, 43 - 47
2. Xu, E.-s .; Lammert, PE; Crespi, VH Nano Lett. 2015, 15, 5124 - 5130
3. Li, X .; Wang, T .; Duan, P .; Baldini, M .; Huang, H.-T .; Chen, B .; Juhl, SJ; Koeplinger, D .; Crespi, VH; Schmidt-Rohr, K .; Hoffmann, R .; Alem, N; Guthrie, M .; Zhang, X .; Badding, JV Am. Chem. Soc. 2018, 140, 4969 - 4972
4.Chen, B .; Wang, T .; Crespi, VH; Badding, JV; Hoffmann, R.Chem. Theory Comput. 2018, 14, 1131–1140
5. Zhan, H .; Zhang, G .; Tan, VBC; Cheng, Y .; Bell, JM; Zhang, Y.-W .; Gu, Y. Nanoscale 2016, 8, 11177 & ndash; 11184