Not only introduction of graphene carbon nanotubes comes, but also new carbon nanomaterials and their auxiliary mechanisms!
Fullerene, carbon nanotubes (CNTs, Carbon Nanotubes) and graphenes (Graphene) are popular carbon nanomaterials in recent years. Currently, five scientists have won the Nobel Prize in this field. Why are carbon nanomaterials widely sought after? For example, bicycles made of carbon fiber-added steel are only a fraction of the weight of ordinary bicycles because of the very small mass of carbon atoms and the chemical bonds between carbon atoms or between carbon atoms and other atoms. Very strong. Therefore, materials mixed with carbon nanometers usually have better mechanical properties and lighter overall weight.
First principles are widely used in physics, chemistry, and materials science. Material design, material prediction, interpretation experiments, etc. are inseparable from the first-principles calculation, because the first principle starts from the Schrödinger equation and requires very few parameters to calculate most of the material properties of the material very accurately; Further combined with the adiabatic assumption, it can also be used to simulate molecular dynamics. In the field of carbon nanomaterials, first-principles calculations are widely used because the electronic correlation of carbon atoms is very weak, and the first-principles calculations can often make very accurate predictions.
This article will introduce some new types of carbon nanomaterials that differ slightly in the way carbon atoms are combined and arranged in well-known fullerenes, carbon nanotubes, and graphene. These subtle differences can be reflected in the final material properties but can vary greatly. A small difference in the arrangement of carbon atoms can translate into large differences in material properties, which is where carbon nanomaterials attract many materials scientists, physicists, and chemists.
1.Hybridization and dimension
There are two main ways to hybridize carbon atoms to carbon nanomaterials: sp2 or sp3. In the sp2 hybrid mode, each carbon atom forms three molecular orbitals uniformly distributed in a plane at an angle of 120 degrees, and an out-of-plane p-orbit, commonly known as pz orbital; the most typical carbon nanomaterials It is a famous graphene. In the sp3 hybrid mode, each carbon atom forms four molecular orbitals that are evenly distributed in space, roughly forming the shape of a regular tetrahedron from the body to the four vertices. A typical solid material represents a diamond, but A typical representative of the world of nanomaterials is Adamantane. Adamantane is a representative of a whole family of materials, and a molecule contains a core of the diamond structure. If it contains multiple cores of diamond structure, then this family of materials will become Diamondoid. Figure 1: Typical carbon nanomaterials classified according to hybridization (sp2, first row; or sp3, second row) and material dimensions.
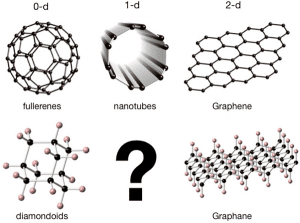
The above is just hybridization, or rather, a mainstream choice that a single carbon atom can make when forming a nanomaterial. When many carbon atoms are combined, in addition to hybridization, they can choose to expand in any direction. Is it a zero-dimensional material or a high-latitude material? The above chart 1 lists various representative materials according to hybridization and dimension.
One-dimensional materials in sp3 hybrid mode lack a typical. Readers familiar with relevant research may think of Polyethylene, but in terms of individual molecules, polyethylene molecules lack some long-range configuration rules, or long-range order, and lack the cravings usually in carbon nanomaterials. Mechanical strength.
2.carbon nanowires
Looking at the material below, is it a bit interesting? Is it solid or macromolecule?
This new type of carbon nanomaterial is both a sp3 hybrid of carbon atoms and a one-dimensional composition of carbon atoms. At the same time, their cross sections are not like a traditional linear organic molecule, but have multiple chemical bonds. Pass through the cross section. This means that these materials are close to diamond insulators in terms of electronic properties. They are far superior in mechanical properties to traditional linear organic molecules, and their mechanical strength is close to that of carbon nanotubes or graphene. Theoretical calculations do confirm these [1], they are called carbon nanowires, or diamond nanothreads.
Is this new material with a strange shape just a theoretical expectation, or can it be actually prepared? It seems that such materials need to start from the synthesis of small organic molecules, after a small to large process, but experimentally [2] is through a process from large to small, starting from the solid state of benzene, after 25GPa high pressure The role of the original sp2 hybrid chemical bond becomes a sp3 hybrid chemical bond under high pressure, thereby transforming the three-dimensional molecular crystal into a one-dimensional carbon nanomaterial.
Long-range ordered one-dimensional nanowires are shown in the example of Figure 2; unordered structures may often be obtained in actual experiments. This figure shows a disordered structure and the results of scanning tunneling microscopy of carbon nanowire crystals obtained in experiments.
3.Applying first-principles calculations
First-principles calculations perform well in predicting the properties of materials. Combining experimental results often leads to more in-depth perspectives on the interpretation of experimental results. In the synthesis of diamond carbon nanowires, due to the harsh experimental conditions, the high pressure of 25GPa needs to be realized in a very small diamond anvil cell (DAC), so the experimental synthesis of materials lacks long-range order, experimental results At first glance, there is a lot of disorder interference. The theoretical calculations can help us distinguish whether the composition contains the new materials we expect.
In theory, we have become a carbon nanowire structure. After adding a certain disorder by introducing the Stone-Wales chemical bond rotation, we can use the theoretical calculation to do the atomic position relaxation and then obtain the optimal structure with the lowest energy. Accurate theoretical calculations can give the distance between atoms in a material, or calculate the radial distribution function in a material. Comparing the theoretical results with the experimental results in Figure 4. It not only confirms that the experimental composition is in agreement with the theoretical structure, but also discerns which atomic structures correspond to the peak resolution of the experimental results.
Figure 4. Comparison of the radial distribution function (RDF) of experimentally synthesized nanowires with the simulated radial distribution function of theoretically generated carbon nanowire structures.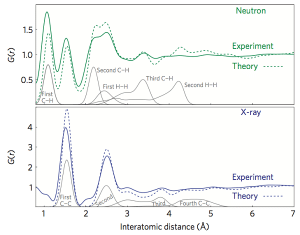
The first principle calculation gives the optical properties of the material. Raman spectroscopy is often a reliable means of characterizing experimental compositions because it does not have to destroy the experimental composition, and spectral peaks can tell us what molecular vibrational modes have Raman activity. One method of calculating the Raman spectrum by density functional theory is to first calculate the dielectric constant of the molecule, and then perform a small displacement of the atom position along the eigenmode of the molecular vibration to calculate the change of the dielectric constant. With the advanced computing power of modern computers, we can now easily calculate the Raman activity of a molecule to determine which structural units are present in the experimental composition. Figure 5 shows a characteristic structural unit included in the synthesis results of carbon nanowires by calculation and analysis of Raman spectroscopy.
Figure 5. Comparison of experimental Raman spectra of carbon nanowires with theory.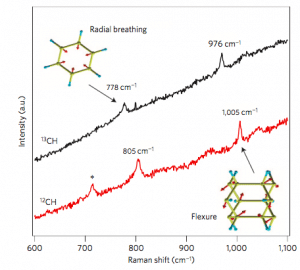
4. Functionalization
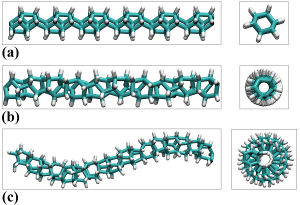
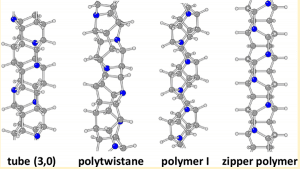
An important feature of carbon nanomaterials is the ability to add various functional groups to them. As long as some small organic molecules are replaced in the preparation stage of the synthetic preparation. In the carbon nanowire material, a simple method involves replacing the hydrogen atom (H) in the reactant with a chlorine atom (Cl), or replacing the carbon atom therein with a nitrogen atom (N) and a boron atom (B). It can be functionalized to change its electronic properties, phonon properties, thermal properties or mechanical properties. Figure 6 shows several typical nanowire structures formed by replacing hydrocarbon groups with nitrogen atoms [4].
The study of replacing benzene with an initial reactant containing a nitrogen atom to synthesize nanowires is published in the article [3]. This replacement is a complete replacement instead of doping, using pyridine (pyridine, C5NH5) instead of the benzene ring to participate in the reaction, the reaction process is still similar to the use of high pressure diamond ballast, the sp2 hybrid carbon is converted into sp3 hybrid carbon And complete the transformation of small molecules into one-dimensional materials.
Using the principle of first principles, we can study by two methods, in which the carbon nanowire material of that structure is synthesized. One is to compare the characterization properties of all candidate structures with experiments, such as Raman spectroscopy, XRD, and so on. The other is naturally sorted by their energy. In calculating the energy of carbon nanowires, their molecular structure and periodicity must be optimized first. However, this one-dimensional material has a characteristic that they have a helical structure, which creates some difficulties in calculation.
If you replace the macromolecules that are truncated at both ends, the energy calculation must be inaccurate; if you use periodic boundary conditions, how do you determine the helix angle? A feasible trick is to select several helix angles for calculation [2]. Each angle is different, which means that the length of a structural repeat period is different along the one-dimensional structure. After calculating a number of different helix angles, the average energy per structural unit (or average per atom) is obtained, and a simple quadratic regression fit is performed on the helix angle. The implicit assumption of quadratic regression fitting is that the effect between two adjacent structural elements is approximately spring-like. Although this is not a completely true hypothesis, it can still capture the main force between adjacent units, because in carbon nanomaterials, covalent bond forces between adjacent atoms and adjacent structural units are used. The Hooke’s law of the spring is approximate.
Figure 6. Four typical diamond carbon nanowires decorated with nitrogen atoms from the literature[4]
5.Mechanical strength
Carbon nanomaterials have a lot of wonderful electrical properties, but now they are widely used in their mechanical lightness: light atoms, strong bonding. Carbon nanowires have the basic unit of diamonds. Will they also have enough strength? Simply put, yes. As shown in Figure 7, the calculations show that the carbon nanowires have a Young’s modulus between 800 and 930 GPa, which is comparable to natural diamonds (1220 GPa). Of course, the mechanical strength of this one-dimensional material is directional. This is both a disadvantage and an advantage: this material concentrates all mechanical strengths in one direction. Some even imagine that this carbon nanowire can be used to make a cable for a space elevator.
Figure 7. Young’s modulus of three different types of diamond carbon nanowires from reference [5].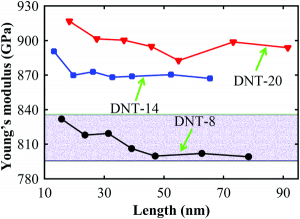
6.Conclusion
Diamond carbon nanowires have recently joined the large family of carbon nanomaterials with a strict one-dimensional structure and high mechanical strength. In the research process, with the help of powerful computing power, through the first-principles calculation, the possible carbon nanowire atomic molecular structure can be studied, and the interpretation of experimental results can be assisted, and the experimental results can be analyzed in depth. Carbon nanowires, as well as many other interesting new features of carbon nanostructures, are waiting for more theoretical calculations and experimental verification to explore.
References
1.Fitzgibbons, T.C.; Guthrie, M.; Xu, E.-s.; Crespi, V.H.; Davidowski, S.K.; Cody, G.D.; Alem, N.; Badding, J.V. Mater. 2014, 14, 43 – 47
2.Xu, E.-s.; Lammert, P.E.; Crespi, V.H. Nano Lett. 2015, 15, 5124 – 5130
3.Li, X.; Wang, T.; Duan, P.; Baldini, M.; Huang, H.-T.; Chen, B.; Juhl, S.J.; Koeplinger, D.; Crespi, V.H.; Schmidt-Rohr, K.; Hoffmann, R.; Alem, N.; Guthrie, M.; Zhang, X.; Badding, J.V. Am. Chem. Soc. 2018, 140, 4969 – 4972
4.Chen, B.; Wang, T.; Crespi, V.H.; Badding, J.V.; Hoffmann, R. Chem. Theory Comput. 2018, 14, 1131 – 1140
5.Zhan, H.; Zhang, G.; Tan, V. B. C.; Cheng, Y.; Bell, J.M.; Zhang, Y.-W.; Gu, Y. Nanoscale 2016, 8, 11177 – 11184